See More Features
See More Features

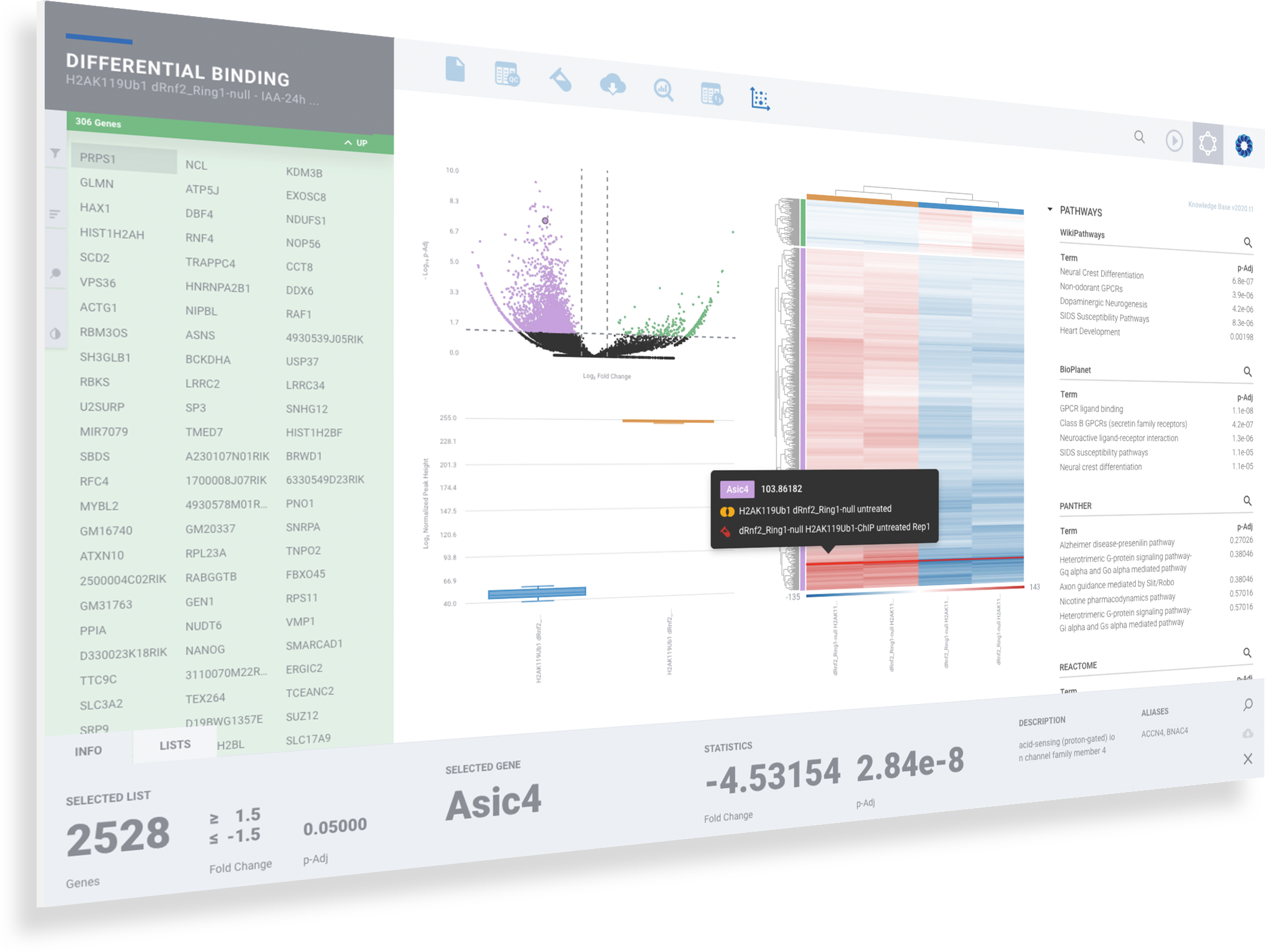
Explore differential Protein Binding visually and interactively
Seamlessly sift and sort through differential promoter accessibility by gene or top pathway. Change cut-offs with new filters. Validate gene signatures and discover new signatures.
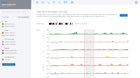
Dynamic Volcano and MA Plots
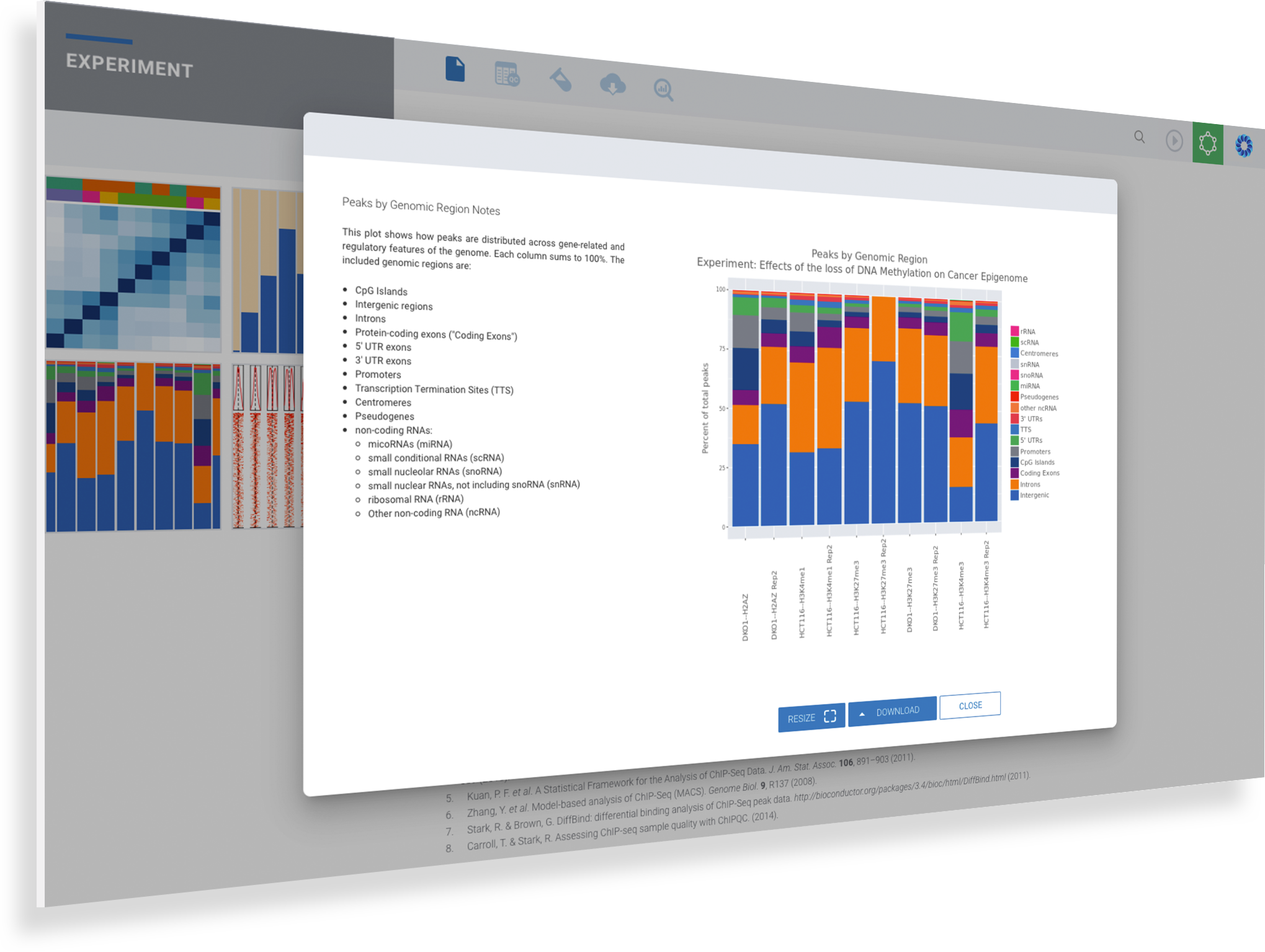
View Protein Binding and Histone Modifications Across Samples with Box & Bar Plots
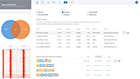
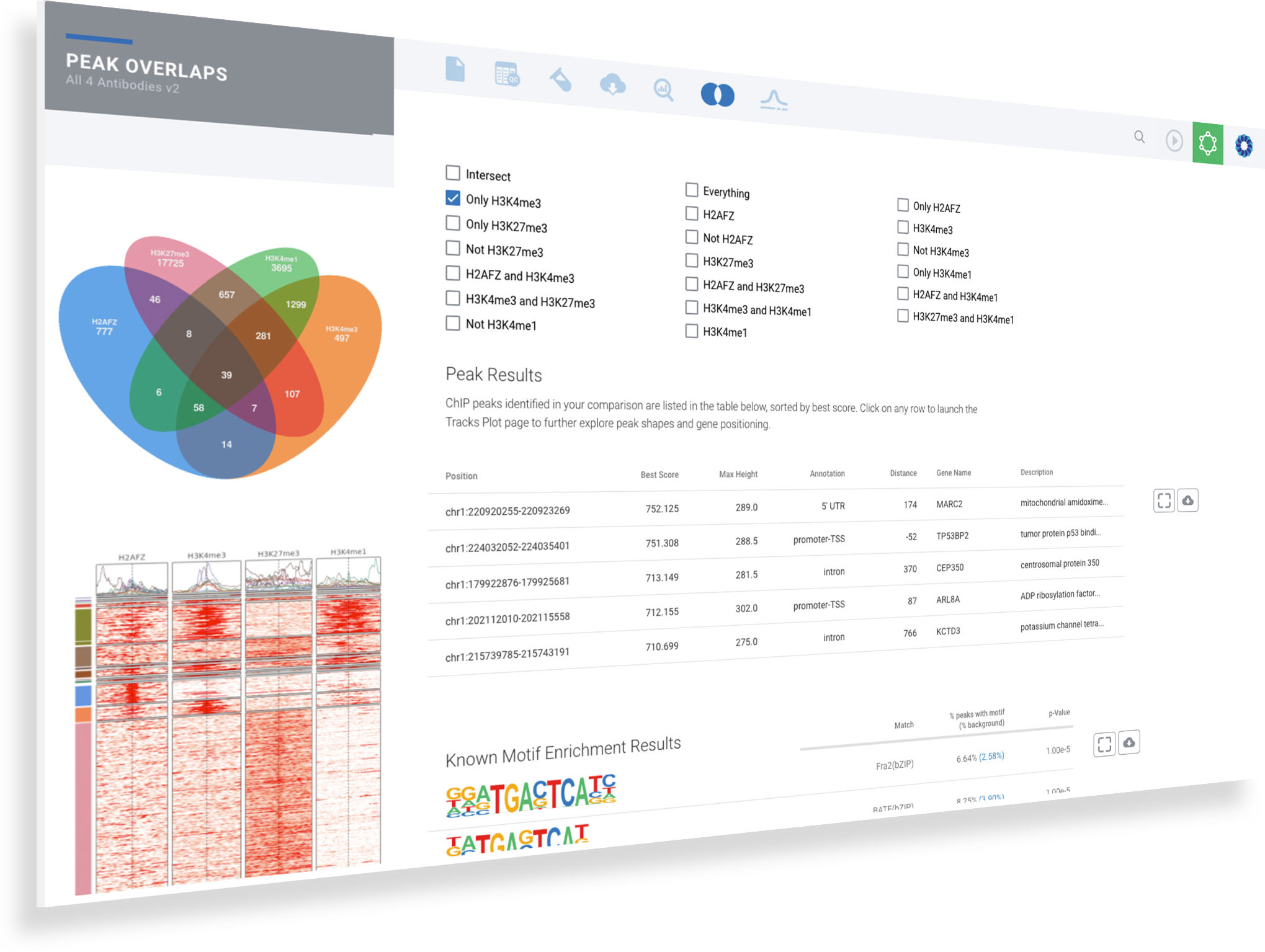
Explore overlaps in protein binding regions.
Using the Peak Overlap interactive analysis, identify unique and overlapping protein binding regions across samples and comparison groups.
Select your samples intersections based on the Venn diagram
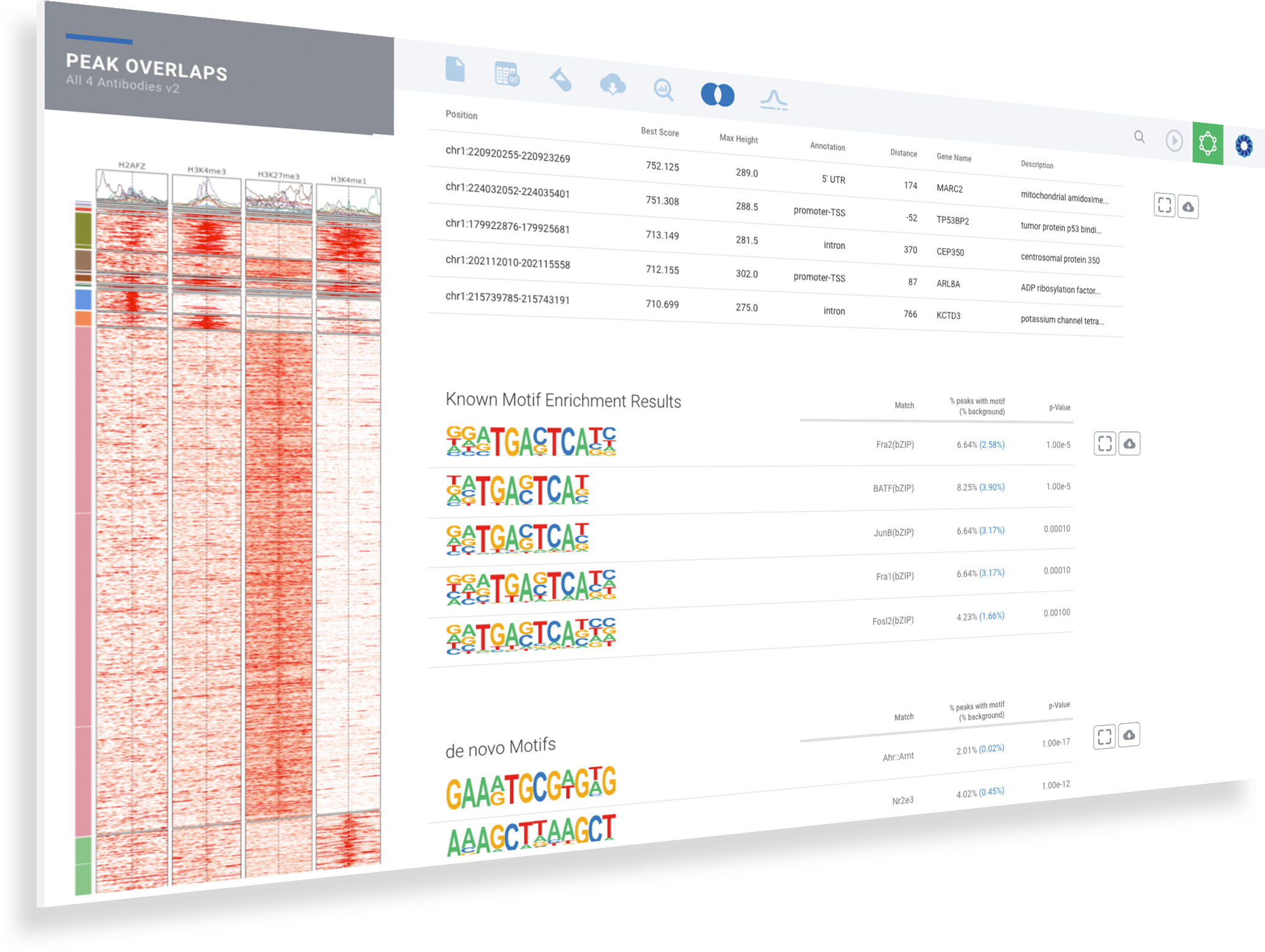
Explore the most significant protein binding regions in the annotated table
Select a meta-analysis to begin exploring the results
Identify common and de novo motifs in accessible sequences
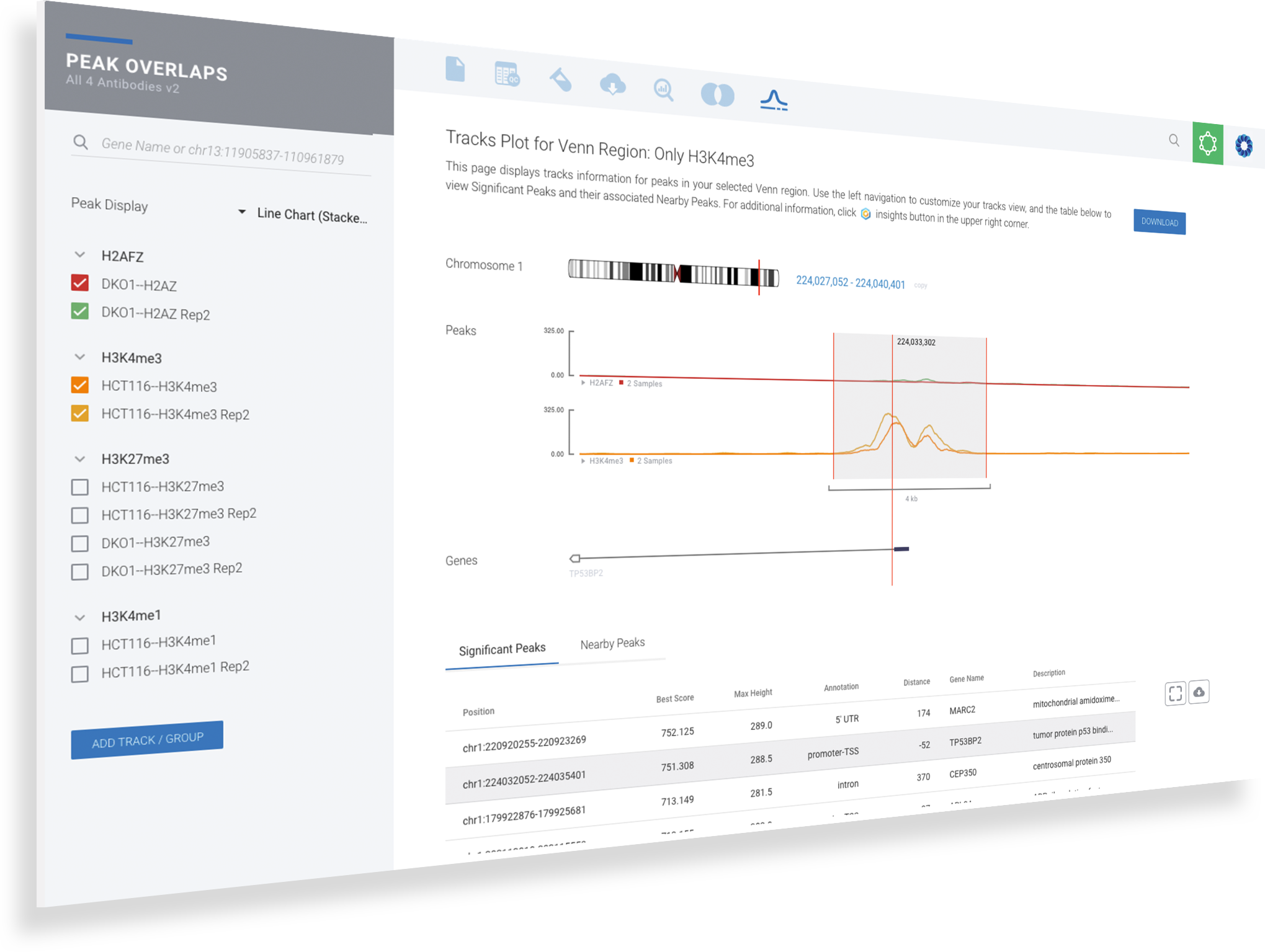
Visualize open chromatin regions.
Locate areas across samples using the integrated genome browser as well as the gene models annotations. Save the time, complexity and the inconvenience of exporting your data to UCSC or IGV.
Search by gene or chromosomal location
Organize samples by groups and select which tracks to display
Advanced platform capabilities inside a simple to use dashboard
Explore your data immediately and stop waiting for results. Seamlessly create new filters to experiment with cut-off values while your interactive plots and interpretation are updated in moments.
Create unlimited filters with different levels of promoter accessibility and significance
Explore downstream genes and enriched pathways
Dive deeper into the pathways and other knowledge bases.
Pathways are shown and sorted by significance. Review the number of genes in each terms, including totals identifying opening or closing of the chromatin on their proximal promoters
Change to any ROSALIND knowledge bases with one click
Navigate the relationship between genes and pathways
Click the orange magnifier to access annotated pathways diagrams
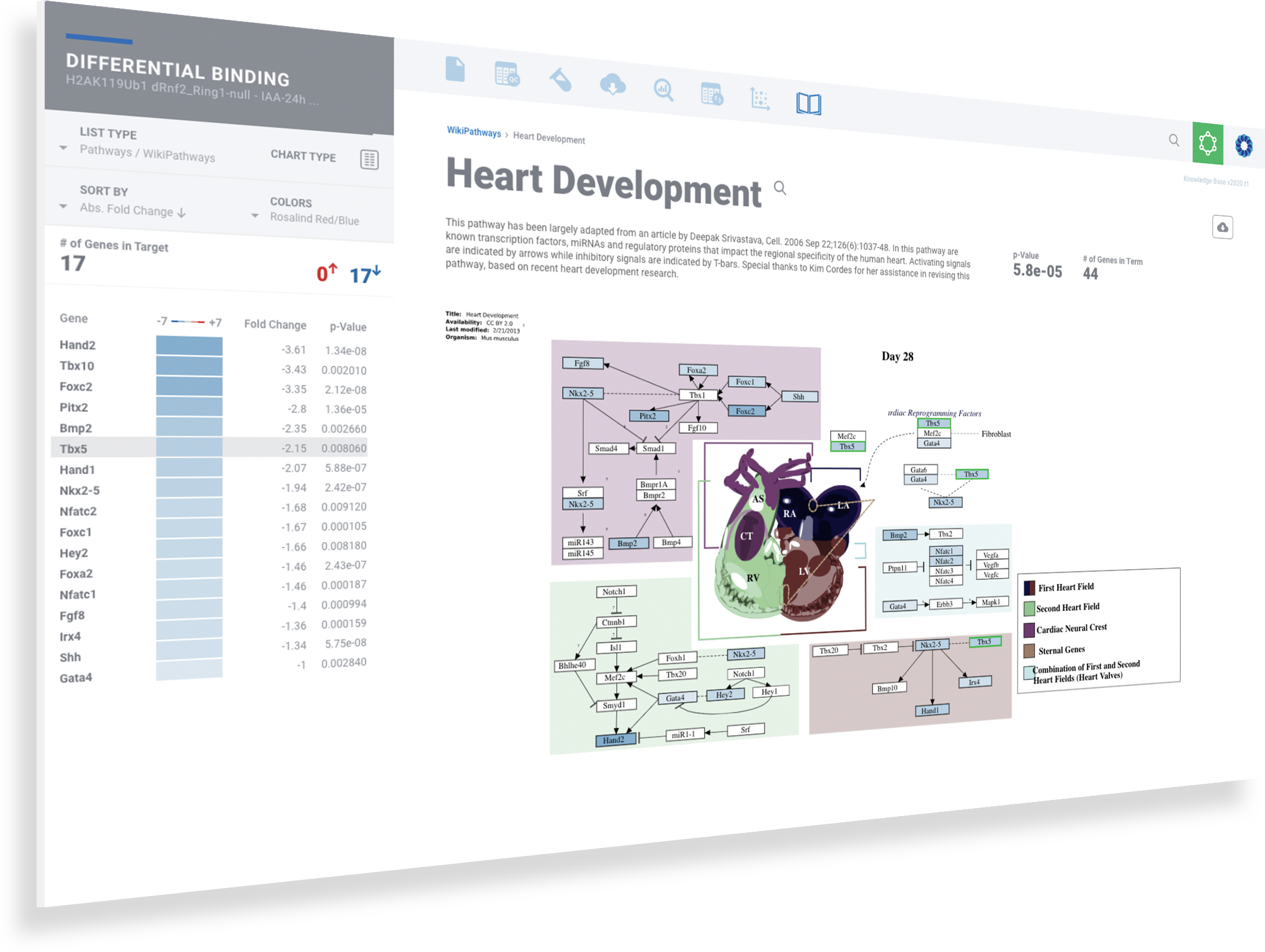
Access rich pathway diagrams
Experience pathways diagrams with detailed descriptions, annotated accessibility change colors and gene heatmaps.
Interact with the pathway diagram to see corresponding genes highlight on the left
Interact with the gene list to see the corresponding genes highlight in the pathway diagram
Access external reference
Download publication-ready pathways diagrams in preferred colors





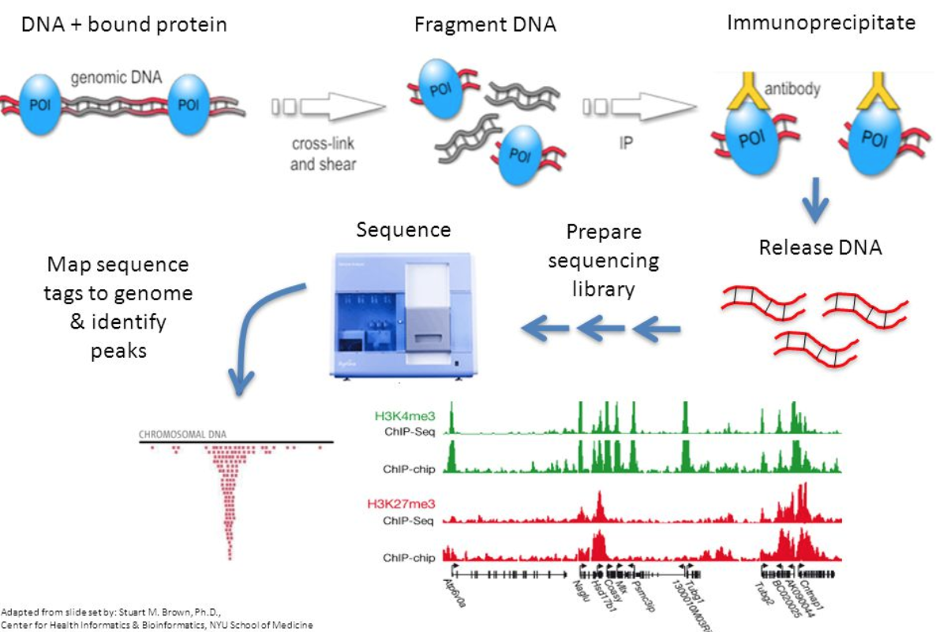
ChIP-sequencing, also known as ChIP-seq, is a method used to analyze protein interactions with DNA. ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. Previously, ChIP-on-chip was the most common technique utilized to study these protein–DNA relations.
ChIP-seq is primarily used to determine how transcription factors and other chromatin-associated proteins like histone marks influence phenotype-affecting mechanisms. Determining how proteins interact with DNA to regulate gene expression is essential for fully understanding many biological processes and disease states. This epigenetic information is complementary to genotype and expression analysis.
Specific DNA sites in direct physical interaction with transcription factors and other proteins can be isolated by chromatin immunoprecipitation. ChIP produces a library of target DNA sites bound to a protein of interest in vivo. Massively parallel sequence analyses are used in conjunction with whole-genome sequence databases to analyze the interaction pattern of any protein with DNA or the pattern of any epigenetic chromatin modifications like histone marks. This method can be applied to the set of ChIP-able proteins and modifications, such as transcription factors, polymerases and transcriptional machinery, structural proteins, protein modifications,histone marks and DNA modifications.[3] As an alternative to the dependence on specific antibodies, different methods have been developed to find the superset of all nucleosome-depleted or nucleosome-disrupted active regulatory regions in the genome, like DNase-Seq, FAIRE-Seq or ATAC-seq.
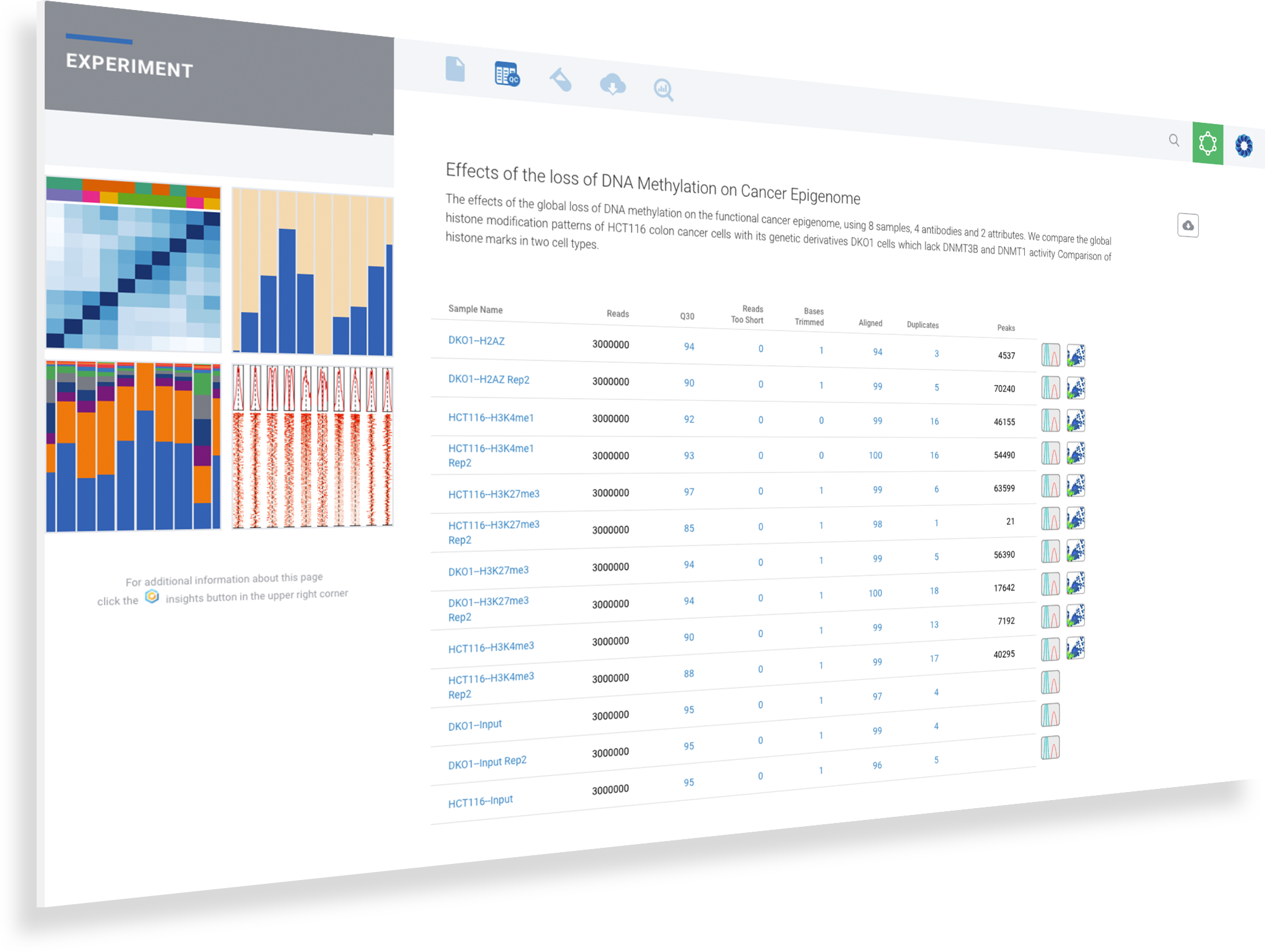
ROSALIND is a cloud platform that connects researchers to experiment design to quality control, peak overlaps, differential binding and pathway exploration in a real-time collaborative environment.
Scientists of every skill level benefit from ROSALIND since no programming or bioinformatics are required. By accepting raw FASTQ sequence data as well as processed counts data, ROSALIND enables powerful downstream analysis and truly insightful visualizations on gene expression datasets. Receive same-day results with every experiment in an interactive experience designed for ease of use and saving valuable time.






I am not a bioinformatician. Can I really perform my own analysis?

Absolutely and other scientists just like you run their own analyses on ROSALIND every day. To learn more how to get started, check out the ROSALIND Quick Start Guide here.
Can the API be used to add experiments to a Space?
Yes, API integration enables production informatics teams to centrally process and distribute results within Spaces to each program or project team requesting an analysis. This is a best practice among pharma R&D teams. API integration also includes Single-Sign-On (SSO) support. Contact sales to learn more sales@onramp.bio
What types of experiments are supported?
The ROSALIND Gene Expression discovery experience supports RNA-seq, NanoString gene and protein panels, and Micro-Array (via counts). Other analysis types include Single Cell, smallRNA-seq, ATAC-seq, and ChIP-seq. We are constantly enhancing our platform and more analysis types are on the way.
Can I download my results and plots?
Yes. All plots, diagrams, source and results files are downloadable on ROSALIND. Look for the Download buttons to access publication-ready figures as well as to download all experiment datasets.
What types of input files are supported?
For Gene Expression experiments, FASTQ files and count files are supported. Compressed FASTQs will have faster upload times. Supported file types: .FASTQ, .FASTQ.GZ, .CSV, .TXT, .RCC (NanoString only)
How do I register for a ROSALIND account and is it free?
What is an Analysis Unit and how is it used on ROSALIND?
Samples that are processed on ROSALIND require an Analysis Unit to unlock the ROSALIND discovery experience. Analysis Units are already included in most subscriptions on ROSALIND. Additional Analysis Units may be purchased in packs of 10 or 50 from the ROSALIND Store. Analysis Units do not expire. A current subscription is required to utilize Analysis Units. Enterprise Subscriptions provide additional flexibility for high-volume environments. Please contact sales to learn more sales@onramp.bio
What is considered a Sample?
Any sample that is prepared for processing on an instrument is considered a Sample for ROSALIND. If a Scientist takes two (2) aliquots of an original sample to have replicates and prepares a library for each, this would be considered two (2) Samples on ROSALIND. On the other hand, a Sample may have multiple files associated with it, depending on how sequencing is performed. A single sample may be single-end, paired-end, and also multi-lane and will still be considered as one (1) Sample.